Manual de tecnologías de medición de concentración de gases y material particulado en chimeneas y atmósfera. Parte 11
3 Cromatografía iónica
Es un tipo de cromatografía líquida (ver Apartado 2) cuyo nombre fue acuñado para describir el análisis de iones disueltos por cromatografía de intercambio iónico, difiriendo en la naturaleza de la resina de intercambio iónico. Los procesos de intercambio iónico se basan en los equilibrios de intercambio entre los iones de una disolución y los iones del mismo signo que están en la superficie de un sólido de elevada masa molecular y esencialmente insoluble.
La columna consiste de un corazón de polímero neutro de alrededor de 10 mm de diámetro.
Dependiendo de si será usado para la separación de iones o cationes, el corazón es ligeramente sulfonado o aminado, lo cual lleva en el primer caso a la formación de una fina cascara superficial exterior de ácido sulfónico (-SO3 -H*), un ácido fuerte, o en algunos casos ácido carboxílico (-COO-H*), y en el segundo caso a la formación de una fina cáscara exterior de grupos de amina primaria (-NH3 +(OH-), o también grupos de amina cuaternaria (-N(CH3)3 +HO-). Esto produce la tradicional resina de intercambio iónico. El hecho nuevo es la introducción de espuma polimérica, aminada o sulfonada y con 100 a 300 nm de diámetro, las cuales son vinculadas electrostáticamente sobre la superficie del corazón del polímero.
4 Detector de ionización de llama
La técnica de medición por ionización de llama se basa en la medición de los iones de una muestra de gas determinado que son formados durante la combustión de hidrógeno. En una cámara de ionización, la nube de iones formados es extraída aplicando un campo eléctrico, vía electrodos, produciendo una corriente eléctrica. Esta corriente es aproximadamente proporcional al caudal de masa del gas, proporcional a su concentración. Es necesario realizar una calibración con muestra patrón.
El detector de ionización de llama consiste de una cámara de combustión (ver Figura 5.6- 4). A través de una boquilla o tobera se introduce hidrógeno puro (combustible), y por un tubo que rodea la tobera aire atmosférico (oxidante) para la combustión. La llama de hidrógeno (2.660 °C) produce una pequeña densidad de iones (valor cero) en la ausencia de la muestra de gas. Los electrodos necesarios para la extracción de la nube de iones son dispuestos cerca de la llama. La misma tobera puede ser usada como uno de los electrodos.
Se emplea un amplificador sensible a corriente continua para lograr una señal de amplitud adecuada. Por una entrada adicional se introduce en la tobera el gas de muestra. Para mediciones continuas se debe mantener constante la temperatura y el caudal de la muestra de gas.
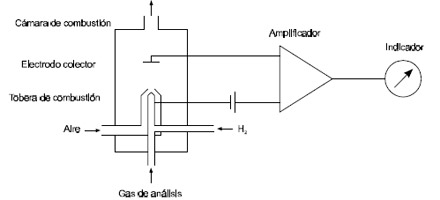 Figura 5.6-4: Detector de ionización de llama |
5 Detector de fotometría de llama.
Se trata de un detector selectivo que se emplea fundamentalmente para los compuestos que contiene azufre (S) y fósforo (P). La muestra pasa a la cámara de combustión donde a través de una boquilla o tobera se introduce hidrógeno puro (combustible), y por un tubo que rodea la tobera aire atmosférico (oxidante) para la combustión y se genera una llama de hidrógeno de baja temperatura. La llama calienta los compuestos de azufre y fósforo y convierte parte del fósforo en una especie HPO que emite bandas de radiación con longitudes de onda alrededor de 510 y 526 nm y el azufre se convierte en S2, emitiendo en una banda centrada en 394 nm. Las bandas se separan empleando filtros adecuados y se mide sus intensidades con fotómetros.
6 Procedimientos luminiscentes
La luminiscencia es el término aplicado al caso en que las moléculas en análisis son excitadas dando una especie cuyo espectro de emisión suministra información para el análisis cualitativo y cuantitativo. Dentro de estos procedimientos se encuentran la fluorescencia molecular y la quimioluminiscencia molecular.
6.1 Fluorescencia molecular
En la fluorescencia molecular fotones de radiación electromagnética son absorbidos por moléculas, alcanzando estas un estado excitado y retornando luego a su estado fundamental con la emisión de radiación; la transición energética en esta caso no genera un cambio de spin de los electrones. A causa de que los niveles de vibración de ambos estados, fundamental y excitado, son similares, el espectro de fluorescencia es una imagen especular del espectro de absorción. El tiempo de vida de un estado excitado simple es de 10 –9 a 10 –6 segundos y el tiempo de vida fluorescente se encuentra dentro de este intervalo.
Es un método muy usado por su elevada sensibilidad y especificidad. La elevada sensibilidad resulta de la diferencia en longitud de onda entre la radiación excitante y la fluorescente. La elevada especificidad proviene de la dependencia de dos espectros, el de excitación y el de emisión, y la posibilidad de medir la vida media del estado fluorescente.
Para los fluorímetros de filtro, como fuente de radiación se emplea la lámpara de mercurio a baja presión que produce 8 líneas muy intensas entre 254 y 773 nm. Las líneas individuales se pueden aislar con los filtros de interferencia o absorbancia adecuados. Para espectros donde se requiere una fuentes de radiación continua, normalmente se emplea una lámpara de arco de xenón a elevada presión que emite un espectro continuo de 300 a 1300 nm.
Como detectores se emplean tubos fotomultiplicadores dado que la señal es de baja intensidad. También se emplean detectores de diodo alineados.
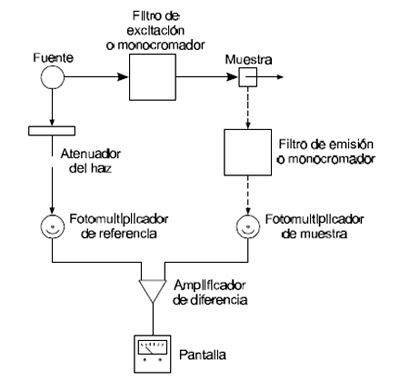 Figura 5.6-5 Esquema de un fluorímetro |
6.2 Método de Quimioluminiscencia
La quimioluminiscencia se produce cuando una reacción química da una especie electrónicamente excitada, la cual emite radiación para volver a su estado fundamental.
La reacción más sencilla para producir quimioluminiscencia se puede formular como
A + B –> C* + D
C* –> C + hn
Donde C* representa el estado excitado de la especie C y hn la radiación emitida. El aire que contiene el contaminante cuya concentración se quiere determinar se mezcla en el reactor con un exceso de un reactivo dado. En general este suele ser ozono. La reacción produce otro gas en un estado excitado que al decaer emite radiación. La intensidad de
esta radiación quimiolumiscencia es proporcional a la concentración del contaminante si el gas auxiliar necesario para producir la reacción está presente en exceso. La instrumentación es muy simple y en general se compone de un recipiente de reacción adecuado y un tubo fotomultiplicador.
El método es relativo y debe ser calibrado con muestras de concentración conocida.
7 Espectrometría de absorción ultravioleta
La absorción de radiación ultravioleta visible está en el intervalo de longitud de onda de 180 a 780 nm. La absorbancia molecular de la radiación ultravioleta es directamente proporcional a la longitud de la trayectoria del haz a través de la muestra y a la concentración de la especie absorbente. Dado que la solución de la muestra que contiene el elemento absorbente debe colocarse en alguna clase de recipiente transparente o cubeta, se produce en este perdidas de radiación por reflexión y dispersión, por lo que la potencia del haz transmitido se debe comparar con la de una cubeta idéntica que solo contenga el disolvente.
El instrumento para medir la absorción de la radiación ultravioleta consta de: 1) fuente de radiación ultravioleta, 2) selectores de longitud de onda, 3) recipiente de muestra, 4) detectores de radiación.
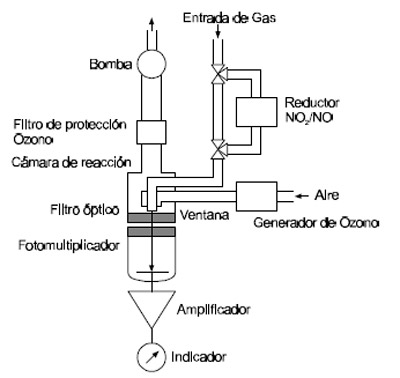 Figura 5.6-6 : Ejemplo del método de medición por quemiluminiscencia |
1) Fuente de radiación ultravioleta : Se necesita una fuente continua cuya potencia no varíe bruscamente en un intervalo considerable de longitud de onda ; se consideran:
a) Lámpara de deuterio e hidrógeno: La excitación eléctrica de deuterio o hidrógeno a baja presión, produce un espectro continuo en la región ultravioleta de 160 a 375 nm.
b) Lámpara de arco de xenón: Se produce una intensa radiación al pasar una corriente a través de una atmósfera de xenón. El espectro continuo es entre 250 y 600 nm.
c) Lámpara de vapor de mercurio con filtros que aíslan la línea de emisión de 254 nm.
2) Recipiente de muestra: Debe ser realizado de un material a través del cual pase la radiación. El cuarzo o sílice fundida permite el paso de radiación por debajo de 350 nm. En la región arriba de 350 nm se puede usar vidrios de silicatos.
3) Detectores de radiación: La mayoría de los espectrofotómetros utilizan tubos fotomultiplicadores.
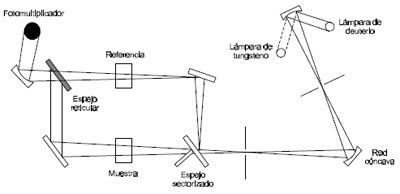 Figura 5.6-7: Esquema de un típico espectrofotómetro manual de doble haz. |
8 Espectrometría infrarroja (dispersiva y no dispersiva)
La región Infrarroja del espectro electromagnético se extiende entre 0,7 y 500 mm. El intervalo más usado es en la región media de 2,5 a 50 mm. La radiación infrarroja no es suficientemente energética como para producir las transiciones electrónicas que se dan cuando se trata de radiaciones ultravioleta y visible. La absorción de radiación infrarroja se limita así en gran parte a especies moleculares para las cuales existen pequeñas diferencias de energía entre los distintos estados de vibraciones y rotaciones. El método se basa en la absorción selectiva de la radiación infrarroja por lo distintos componentes de una muestra.
Al entrar en interacción con la radiación infrarroja, las moléculas absorben porciones de la misma a longitudes de onda específicas. La multiplicidad de vibraciones que ocurren simultáneamente producen un espectro de absorción altamente complejo que es unívocamente característico de los grupos funcionales que integran la molécula y la configuración que incluye a la molécula misma. Luego cuando se interpreta un espectro es posible establecer que están presente cierto grupos funcionales, cierto compuestos de interés, y determinar su concentración por comparación. Parte de la energía de la radiación infrarroja es absorbida por el compuesto en análisis en forma proporcional a su concentración; en una célula de referencia no existe dicha absorción y por diferencia se puede determinar la concentración.
La instrumentación usada para el estudio de espectrometría infrarroja se divide en dos grupos:
a) Instrumentos dispersivos: Usan un prisma o retículo para seleccionar o aislar la longitud de onda deseada.
b) Instrumentos no dispersivos: usan filtros de interferencia, fuentes de láser sintonizable o un interferómetro (espectrometría infrarroja por transformada de Fourier), para aislar la longitud de onda deseada.
En la región infrarroja se usan espejos de primera superficie pues el vidrio o cuarzo usado en las lentes son opacos a esa radiación.
Se necesita una fuente de radiación infrarroja continua. Consisten en un sólido inerte que se calienta eléctricamente a una temperatura entre 1.500 y 2.200 K. La máxima intensidad radiante se produce entre 2 a 1,7 mm. Se pueden mencionar : a) emisor de Nernst, constituido por óxidos de tierras raras, b) fuente global, constituido de varilla de carburo de silicio, c) filamento incandescente, constituido de un filamento de alambre de níquel cobre, d) arco de mercurio, especialmente para infrarrojo lejano (l> 50 mm), e) lámpara de filamento de tungsteno, ideal para infrarrojo cercano (2,5 a 0,78 mm). f) fuente láser de dióxido de carbono, que produce una banda en el intervalo de 11 a 9 mm.
Los detectores usados para longitudes de onda arriba de 1,2 mm se clasifican en dos grupos:
a) Detectores térmicos: la radiación infrarroja produce un calentamiento que altera alguna propiedad física del detector (por eje. expansión de un sólido, gas o fluido, resistencia eléctrica, termocupla, termopila, piroeléctrico).
b) Detectores de fotones: usa el efecto cuántico de la radiación infrarroja para cambiar la propiedad eléctrica de un semiconductor (detector fotovoltaico o fotoconductor).
Las moléculas heteroatómicas como CO y CO2 poseen un espectro de absorción característica típica en el intervalo de radiación infrarrojo. La radiación infrarroja se hace pasar a través de un celda conteniendo el gas de muestra que se desea analizar, y la absorción cuantitativa de energía por el CO es medida por un detector apropiado en un fotómetro no dispersivo.
Para los casos de contaminantes con espectros de absorción muy similares, puede haber interferencias. En ese caso, se puede utilizar una celda en serie con una muestra del gas que no se quiere medir y puede interferir que elimina la parte del espectro correspondiente.
Dependiendo del método de almacenaje de muestra, se distingue entre los métodos de Absorción radiación infrarrojo no dispersivo y Correlación filtro de gas.
8.1 Absorción radiación infrarrojo no dispersivo
Se utiliza un doble haz de radiación infrarroja (ver Figura 5.6-8), que pasan a través de dos celdas; una es la de medición llena con el gas de muestra y la otra, de referencia, llena por ejemplo de N2. En la primera celda se produce la absorción en el componente que se desea medir, mientras que en la de referencia no hay absorción. Una rueda interruptora (“chopper”) permite que el haz llegue alternativamente al detector. El método usa el receptor de luz como almacenaje. El detector consiste en dos comportamientos separados por un diagrama a los que llega la radiación no absorbida en las celdas, llenos con el mismo tipo de gas que se analiza. Este absorbe la energía radiante del haz (mayor en la parte que recibe el haz que pasó por la celda de referencia), aumentando su temperatura y ejerciendo una presión sobre el diafragma La modulación producida por la rueda interruptora (“chopper”) producen variación periódica de presión en las cámaras. Estas señales son detectadas y convertidas en señal eléctrica, o por un capacitor de membrana (el diafragma forma parte del capacitor y la variación de posición da lugar una modificación de su capacidad) o con un detector de micro flujo, los cuales miden el flujo de presión compensado entre las dos cámaras receptoras.
8.2 Correlación filtro de gas
En el método de correlación filtro de gas (ver Figura5.6-9) se usa un sólo haz de radiación infrarroja que pasa por la celda de medición.. Para limitar la sensibilidad del fotómetro a una banda angosta de interés se hace pasar la radiación infrarroja alternativamente por el filtro con CO, produciendo un haz de referencia, y con otro gas, por ejemplo N2, que es transparente a la radiación infrarroja de interés, generándose el haz de medición, que luego es absorbido por el CO de la muestra. Las cámaras de filtrado son alternativamente puestas delante del haz de luz con un sistema rotativo.
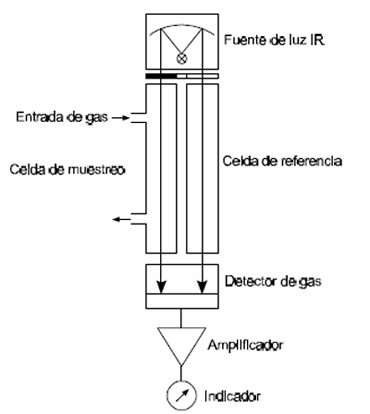 Figura 5.6-8: Absorción de radiación infrarroja no dispersivo |
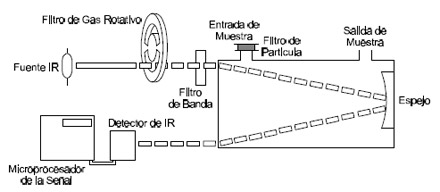 Figura 5.6-9: Método de correlación filtro de gas |
9 Métodos potenciométricos
Los denominados métodos potenciométricos están basados en la medición del potencial de las celdas electroquímicas empleadas cuando no se producen corrientes apreciables. Se emplea para la detección de puntos finales en métodos volumétricos de análisis. También incluyen los métodos en los que las concentraciones de los iones se obtienen directamente del potencial de un electrodo de membrana selectiva de iones.
El equipamiento necesario para el análisis por métodos potenciométricos es simple e incluye un electrodo de referencia, un electrodo indicador (que puede ser metálico o de membrana) y un dispositivo para medir potencial.
9.1 Método de electrodo selectivo de iones o electrodo específico (denominado también electrodo a membrana selectiva de iones)
Desde el punto de vista analítico el método de electrodo selectivo de iones es una herramienta de medición ideal por su habilidad de monitorear selectivamente la actividad de ciertos iones en solución en forma continua y no destructiva. El sistema está compuesto de un electrodo de referencia, una solución interna y una membrana (electrodo indicador o de trabajo, cuya respuesta depende de la concentración del elemento o compuesto en estudio) a través de la cual pasa selectivamente los iones cuya concentración se desea determinar. Si ocurre una separación de cargas entre iones en una interface, se genera una diferencia de potencial a través de la interface. El potencial de un electrodo selectivo de iones está compuesto de dos o más contribuciones discretas que aparecen de los varios procesos en las interfaces y en el volumen o masa del material de la membrana activa. El problema es encontrar una interface cuya composición favorezca selectivamente un tipo de iones sobre otros. En una solución diluida, la actividad de los iones es usualmente cercano a la concentración de iones. El mecanismo general por el que se produce en estos montajes un potencial selectivo de iones es independiente de la naturaleza de la membrana y es completamente diferente del mecanismo que origina el potencial en los electrodos indicadores metálicos. En estos últimos el potencial se produce debido a la tendencia a que tenga lugar una reacción de oxidación/reducción en la superficie del electrodo. En los electrodos de membrana, por el contrario, el potencial observado es una especie de potencial de unión que se forma en la membrana que separa la disolución del elemento o compuesto en estudio y la disolución de referencia.
Los electrodo de referencia deben ser reversibles, presentar un potencial que sea constante con el tiempo, volver al potencial original después de haber estado sometido a corrientes pequeñas y presentar poca histéresis con los ciclos de temperatura Algunos electrodos de referencia son: a) electrodos de calomelanos, que consisten en mercurio en contacto con una disolución saturada de cloruro de mercurio y que también contiene una concentración conocida de cloruro de potasio b) electrodos de plata – cloruro de plata (Ag-AgCl), que consiste en un electrodo de plata sumergido en una disolución de cloruro de potasio (KCl) que ha sido saturada de cloruro de plata.
Los electrodos de membrana se llaman también electrodos de iones debido a la alta selectividad de la mayoría de estos dispositivos. Hay dos tipos de electrodos de membrana selectivos de iones:
1. Electrodo de membrana cristalina
a) Cristal único. Eje. LaF3 para F-.
b) Policristalina o mezcla de cristales.
Eje. Ag2S para S2- y Ag+
2. Electrodos de membrana no cristalina
a) Vidrio Eje. Vidrios de silicatos para Na+ y H+
b) Líquido Eje. Intercambiadores de iones líquidos para Ca2+ y portadores neutros para K+.
c) Líquido inmovilizado en un polímero rígido.
Eje. Matriz de cloruro de polivinilo para Ca2+ y NO3-.
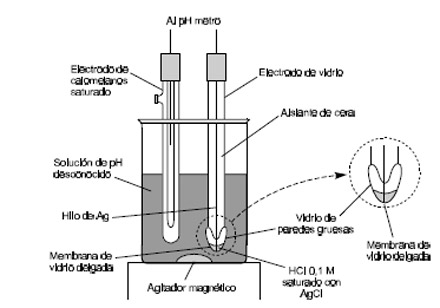 Figura 5.6-10: Sistema de electrodos típicos para medir el pH |
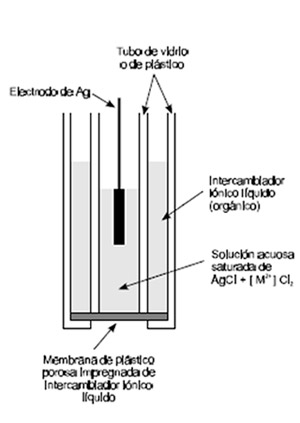 Figura 5.6-11: Electrodo de membrana líquida sensible a M2+ (catión cuya actividad se determina) |
Por: Dr. Jaime A. Moragues
Fuente: blogaustral.org