Viabilidad de un Proceso para la Eliminación conjunta de H2S Y NH3 contenido en Efluentes Gaseosos. Parte 7
 |
Martín Ramírez Muñoz TESIS DOCTORAL |
La presente Tesis ha sido co-dirigida por los Doctores D. Domingo Cantero Moreno, Catedrático de Ingeniería Química y D. José Manuel Gómez Montes de Oca, Profesor Titular de Ingeniería Química de la Universidad de Cádiz, y cumple los requisitos exigidos por la legislación vigente.
Fdo.: Dr. D. Domingo Cantero Moreno Fdo.: Dr. D. José Manuel Gómez Montes de Oca
Fdo.: Dr. D. José María Quiroga Alonso
Director del Dpto. de Ingeniería Química, Tecnología de Alimentos y Tecnologías del Medio Ambiente
Universidad de Cádiz
3.3. ELIMINACIÓN CONJUNTA DE MEZCLAS H2S/NH3 EN AIRE
3.3.1. Estudio del desarrollo de biopelículas mixtas
Para poder llevar a cabo un procedimiento similar al utilizado en la inmovilización por separado de ambas bacterias, se formuló un medio en el cual pudieran crecer ambas bacterias simultáneamente. El principal problema radica en que la bacteria Nitrosomonas europaea presenta inhibición cuando la concentración de iones sodio es alta (Hunik et al., 1992), elemento presente en la fuente de sustrato de la bacteria Thiobacillus thioparus.
Por tanto, se formuló un medio en el cual se disminuyó la concentración de tiosulfato al 50%, debido a que una disminución mayor no se considera viable ya que la velocidad de consumo de tiosulfato por Thiobacillus thioparus es muy alta. También se disminuyó la concentración de amonio de 50 a 30 mM.
La composición en gramos por litro fue: 1,98 g de (NH4)2SO4 (para 30 mM NH4 +), 5,0 g de Na2S2O3, 1,2 g de Na2HPO4, 1,8 g de KH2PO4, 0,1 g de MgSO4·7H2O, 0,03 g de CaCl2, 0,02 g de FeCl3 y 0,02 g de MnSO4.
Se disolvieron todos los compuestos, menos el FeCl3, que se esterilizó por filtración, añadiéndose al medio esterilizado en autoclave (121 ºC, 20 min). El pH final del medio se ajustó a 8,0 mediante la adición de NaOH, conservándose en oscuridad a 4ºC.
Se hicieron ensayos de crecimiento de ambas bacterias por separado, inoculando 90 ml con 10 ml de inóculo para probar la viabilidad del nuevo medio y realizar la inmovilización de forma semejante a la realizada para las bacterias por separado.
3.3.2. Eliminación en dos etapas en serie
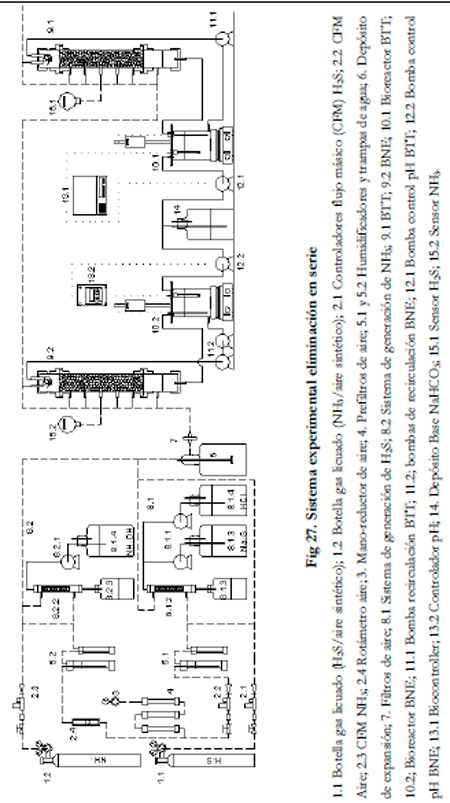
Una vez finalizado el estudio de la eliminación por separado de H2S y NH3, se realizó un experimento para ver si era viable eliminar una corriente contaminada con H2S y NH3, conectando ambos biofiltros en serie. El primer biofiltro fue el de Nitrosomonas europaea (BNE) y el segundo biofiltro el de Thiobacillus thioparus (BTT). Esta disposición se adoptó debido a la alta solubilidad del NH3 en agua, por lo que se hace necesaria la colocación del BNE en primer lugar para que se produzca la degradación del amoniaco en primer lugar, pasando el H2S al BTT para su degradación.
Se trabajó con un tiempo de residencia del gas de 60 segundos, una carga de amoniaco constante de 10 gN m-3h-1, un pH entre 7,5-7,6 para el primer biofiltro y entre 7,4-7,5 para el segundo. Se fue aumentando progresivamente la carga de ácido sulfhídrico a valores de 2,9; 7,2; 11,5 y 15,8 gS m-3h-1. El caudal de recirculación en cada biofiltro fue el mismo que el utilizado en los experimentos anteriores, 308 ml min-1 para el BTT y 445 ml min-1 para el BNE. La concentración de nitrito se mantuvo por debajo de 150 mM y la de sulfato fue inferior a 4 g l-1.
En la Figura 27 se presenta un esquema del sistema experimental utilizado.
 |
3.3.3. Eliminación en dos etapas en serie co-inmovilizando ambos microorganismos
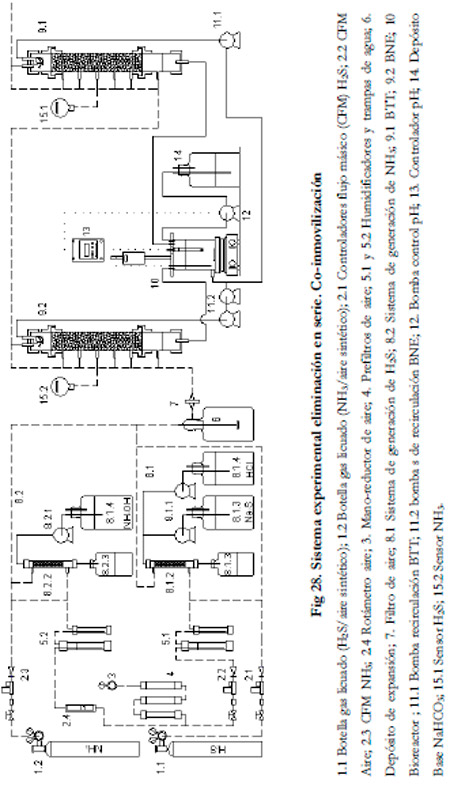
Para crear una biopelícula mixta se optó por utilizar un mismo medio de recirculación de nutrientes. De esta forma, se consigue co-inmovilizar los dos biofiltros con ambos microorganismos, y se dispone del BNE con una biopelícula de Thiobacillus thioparus sobre la biopelícula de Nitrosomonas europaea y, en el BTT, una biopelícula de Nitrosomonas europaea sobre la biopelícula de Thiobacillus thioparus.
Para formular el medio de recirculación se empleó un 50% de cada medio (ATCC 290 y ATCC #2265). Se utilizó un único bioreactor del cual toman las bombas de recirculación los nutrientes necesarios para ambos biofiltros. Como controlador de pH se empleó el Controlador CRISON PH 28.
Se trabajó con un tiempo de residencia de 60 segundos, un pH entre 7,5-7,6, una concentración de nitrito por debajo de 50 mM, una concentración de sulfato menor de 4 g l-1, un rango de cargas para el NH3 desde 1,25 hasta 10 gN m-3h-1 y entre 2,89 y 7,19 gS m-3h-1 para el H2S. El caudal de recirculación se mantuvo constante al igual que en el experimento anterior con unos valores de 308 ml min-1 para el BTT y de 445 ml min-1 para el BNE.
En la Figura 28 se ha representado un esquema del sistema experimental empleado.
 |
3.3.4. Eliminación en paralelo
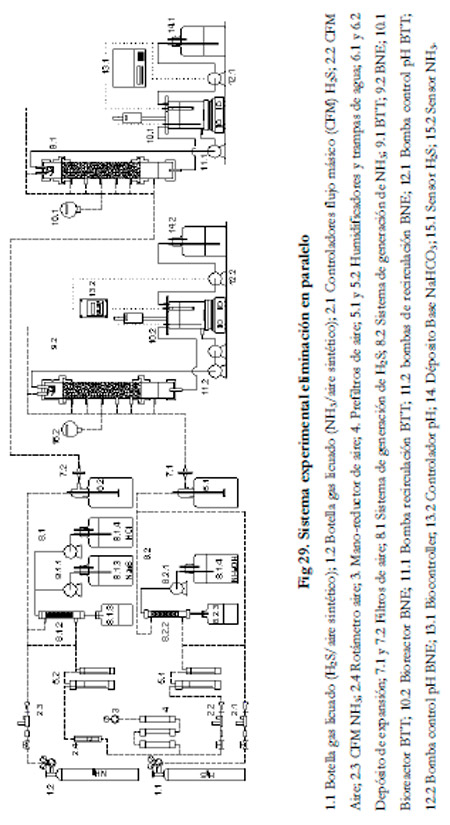
Una vez finalizado el experimento de eliminación en serie, se cambió la configuración de ambos biofiltros para pasar a trabajar en paralelo. De esta forma, se puede ver cuál de las dos biopelículas creadas presenta una mayor capacidad de eliminación al tratar mezclas de H2S/NH3 en aire.
El medio de recirculación empleado fue el mismo que en el experimento anterior, se formuló con un 50% de cada medio (ATCC 290 y ATCC #2265).
Se trabajó con un tiempo de residencia de 60 segundos, un pH entre 7,5-7,6, una concentración de nitrito por debajo de 100 mM, una concentración de sulfato menor de 4 g l-1, un rango de cargas para el NH3 desde 4 hasta 9 gN m-3h-1 y entre 2,89 y 10 gS m-3h-1 para el H2S. El caudal de recirculación se mantuvo constante, al igual que en el experimento anterior, con unos valores de 308 ml min-1 para el BTT y de 445 ml min-1 para el BNE.
Para la generación de la corriente gaseosa se empleó la configuración que se presenta en la Figura 29.
 |
3.4.MÉTODOS DE ANÁLISIS
3.4.1. Determinación de la concentración celular
La medida de la cantidad de biomasa total en los medios de cultivo se realizó mediante recuento en cámara de Neubauer (Figura 30). La cámara de Neubauer posee dos zonas de recuento. En la Figura 31 podemos ver un detalle de la zona o cuadricula de recuento.
 |
Para esta cámara el recuento de una diagonal (4 cuadros) equivale directamente al número de bacterias en millones de células por mililitro.
El número de células óptimo debe estar en el rango de 5 a 12 en cada cuadrado pequeño.
Al menos debe haber 600 células en el volumen determinado por la cámara para que el recuento sea significativo. Además, se recomiendan múltiples llenados de la cámara para promediar la reproducibilidad del llenado.
En casos de muestras en que las bacterias presenten una alta movilidad, se utiliza para hacer la dilución el diluyente de Norris-Powell. Este diluyente se prepara adicionado 5ml de formaldehído a un litro de agua, se ajusta el pH de la disolución con hidrogeno fosfato disódico entre 7,2 y 7,4 y se añade una pequeña cantidad de dodecilsulfato sódico.
Se empleó un microscopio óptico Olympus BH-2 que dispone de 5 objetivos de aumentos x4, x10, x20, x40 y x100, siendo el óptimo para el recuento de x40. La imagen es captada por una cámara analógica y dirigida a un monitor de 14" para su recuento.
3.4.2. Determinación de la biomasa inmovilizada
Para obtener la cantidad de biomasa inmovilizada, se realiza el recuento de bacterias en una unidad de soporte y se divide la cantidad total de biomasa entre el peso del soporte (Gómez et al., 2000; de Ory et al., 2004). Para el recuento de la cantidad de bacterias se toma una unidad de soporte o parte de ella y se elimina el líquido retenido, secando la muestra suavemente con papel absorbente. A continuación, se sumerge en 25 ml de medio estéril y se somete a ultrasonido durante 15 min para provocar la desorción de las bacterias, que permite realizar un recuento de la cantidad total de bacterias mediante la cámara de Neubauer. Para determinar la cantidad exacta de soporte que se ha utilizado en el análisis, se realiza un secado a 80ºC en estufa durante 24h. Esta técnica ha sido validada estudiando la resistencia de las células al ultrasonido y la eficacia de desorción de las células del soporte.
3.4.3. Determinación de biomasa viable
Para realizar el recuento de biomasa viable se utilizó la técnica de siembra en placa mediante la realización de diluciones seriadas. Para la preparación del medio con agar-agar se añadió al medio ATCC 290 (Thiobacillus thioparus) 15 g de agar-agar por litro de medio (1,5% p/v), para el medio ATCC #2265 (Nitrosomonas europaea) se añadió 30 g de agar-agar a la solución 1, de forma que al mezclar las tres soluciones tenemos un 2% p/v.
Cada placa se completó con 20 ml medio. Una vez solidificado, se sembró cada una con 50 μl de cada dilución, por duplicado, incubándose a 30ºC y en caso de siembra de Nitrosomonas europaea en oscuridad.
Al cabo de 48 h se puede apreciar el crecimiento, pero conviene esperar 4-5 días para poder realizar el recuento de las colonias. Para el recuento se procuró utilizar las placas que contenían entre 30 y 300 colonias. El número de unidades formadoras de colonia o UCF/ml de la muestra original será:
 |
Dado que ambos compuestos no se encontrarán presentes de forma simultánea en las muestras, se puede obtener mediante valoración con yodo la concentración de cada uno de ellos. En el caso de muestras con ambos compuestos, se añade una solución de carbonato de cinc que elimina el sulfuro presente, pudiendo obtener de esta forma la concentración de cada especie. El nitrito es una especie que interfiere en la valoración, y en estos casos se añade ácido sulfanílico (C6H7NO3S).
Las soluciones necesarias son:
- Ácido sulfúrico diluido al 10%.
- Solución de yodo 0,0001M: Añadir 20 ml de la disolución de yodato y 20 ml de la disolución de yoduro potásico y enrasar a un litro. Disoluciones:
* Yodato (Calidad patrón primario): Pesar exactamente 0,2675 g (secado a 110ºC) y disolver en 250 ml agua destilada.
* Yoduro potásico: Pesar 2,1 g aproximadamente y disolver en 250 ml de agua destilada. - Engrudo de almidón: Se disuelven 2 g de almidón soluble calidad laboratorio en 100 ml de agua caliente. Se le añaden 0,2 g de ácido salicílico, como conservador.
- Ácido sulfanílico (C6H7NO3S): Diluir 7 g en 500 ml
El procedimiento de análisis consiste en filtrar la muestra con un filtro de 0,45 μm y tomar un volumen conocido para proceder a su valoración. Añadimos 1 ml de ácido sulfúrico al 10%, 4 ml de ácido sulfanílico (solo si hay nitritos) y 5 gotas de almidón. Se añade la disolución de yodato hasta el viraje a azul de la muestra.
 |
3.4.5. Concentración de sulfato
El método empleado para la determinación de sulfato fue una modificación del método turbidimétrico clásico (Clescerl et al., 1989). Este método se basa en la reacción que se produce entre el cloruro de bario, en medio ácido, y el sulfato, formándose un precipitado blanco de sulfato de bario. Para conseguir que el precipitado de sulfato de bario se mantenga en suspensión se utiliza una disolución acondicionadora que contiene glicerina y alcohol, de forma que se modifica la viscosidad de la muestra, permitiendo así una turbidez estable. La medida de esta turbidez a 420 nm se relaciona con la concentración de sulfato.
Para llevar a cabo este método de análisis se necesitan una serie de patrones que se detallan a continuación:
- Solución madre de 1000 ppm de SO4 =: Disolver 1,479 g de Na2SO4 anhidro secado a 110ºC durante 2h, en agua destilada hasta 1 litro. 1 ml de esta solución equivale a 1 mg de SO4 =.
- Solución patrón de SO4 = (100 ppm): Disolver 100 ml de la solución madre en 1 litro de agua destilada. 1 ml de esta solución equivale a 0,1 mg de SO4 =.
Los reactivos necesarios son los siguientes:
- Solución ácida acondicionadora: Añadir 10 ml de glicerina a una disolución que contenga 6 ml de HCl concentrado, 60 ml de agua destilada, 20 ml de alcohol etílico y 15 g de cloruro de sodio.
Para construir la recta patrón, se preparan los patrones para realizar la recta de calibrado desde 5 hasta 100 ppm de sulfato. A continuación se toman 10 ml de cada patrón y del blanco; se adicionan 0,4 ml de solución ácida acondicionadora, más la punta de una espátula de BaCl2 2H2O. Se agita en vortex durante 1 minuto y se deja reposar otro minuto midiendo la absorbancia a 420 nm dentro de los 2 minutos siguientes.
El tratamiento de las muestras se realiza con una filtración previa de la muestra por un filtro de 0,22 μm. A continuación se prepara la dilución necesaria de la muestra hasta un volumen final de 10 ml y se trata de forma análoga a los patrones.
3.4.6. Análisis de acido sulfhídrico
Para el análisis del ácido sulfhídrico en fase gas se empleó un sensor específico de la marca Crowcon (Model GASFLAG, TXGARD-IS). Este sensor da una relación lineal entre la concentración de H2S y el voltaje de respuesta del sensor (medido con voltímetro). En ausencia de H2S, el voltaje es de 40 mV y para concentraciones mayores se puede regular el sensor fijando para cualquier concentración un valor máximo de 430 mV. El coeficiente de regresión lineal (r2) para todas las calibraciones fue mayor de 0,99. Para la calibración más sensible la variación de ±1mV equivale a ±0,19 ppmv de H2S.
3.4.7. Concentración de amoniaco
Para determinar la concentración de amoniaco en medio líquido se utilizó un método colorimétrico, concretamente la nesslerización directa (Clescerl et al., 1989). El reactivo Nessler (I2Hg 2IK) reacciona con el amonio originando un compuesto pardo rojizo o amarillo, dependiendo de la concentración de amonio, que permite la determinación colorimétrica del ión amonio.
Las soluciones necesarias son:
- Solución madre de nitrógeno amoniacal (1000 mg NH3 l-1): Disolver 3,147 g de NH4Cl secado a 100ºC, en agua destilada exenta de amoniaco hasta 1 litro (1 ml de esta solución equivale a 1 mg de NH3).
- Solución patrón de nitrógeno amoniacal (10 mg NH3 l-1): Diluir 10 ml de la solución madre hasta 1 litro con agua exenta de amoníaco. (1 ml equivale a 0,01 mg de NH3).
- Solución NaOH 6N (24g de NaOH en agua destilada hasta 100 ml)
- Solución de ZnSO4 al 10%.
- Solución de Sal de Rochelle: Disolver 50g de tartrato sódico potásicotetrahidratado C4H4O6KNa 4H2O en 100 ml de agua exenta de amoníaco.
- Nessler: Reactivo comercial de Hach (Nessler Reagent 2119432).
El reactivo debe producir el color amarillo característico con 0,1 mg de NH3 l-1 dentro de los diez minutos siguientes a su adición y no debe producirse precipitado con pequeñas cantidades de amoniaco en un intervalo de tiempo de dos horas.
Para la construcción de la recta patrón, se preparan patrones desde 0,5 hasta 6 ppm de NH3. Se toman 5 ml de cada patrón y del blanco, se le adicionan 0,1 ml de la solución de sal de Rochelle (para evitar la precipitación de los iones Ca2+ y Mg2+ presentes), 0,1 de Nessler y se homogeneiza. Después de un tiempo de reacción de 20 minutos, se mide la absorbancia de la solución a 425 nm.
El tratamiento de las muestras se realiza con una filtración previa por un filtro de 0,22 μm.
A continuación se prepara la dilución necesaria de la muestra hasta un volumen final de 10 ml, y se le añaden 0,1 ml de solución de sulfato de cinc. Tras agitación, se agregan 50 μl de NaOH 6N para obtener un pH cercano a 10,5. Se deja reposar para que sedimente el precipitado, debiendo quedar el líquido sobrenadante incoloro y transparente, aunque se puede centrifugar, si es necesario.
Se toman 5 ml del líquido sobrenadante, a los que se añaden 0,1 ml de solución de sal de Rochelle, 0,1 ml de Nessler y se homogeneiza. Después de un tiempo de reacción de 20 minutos, se mide la absorbancia de la solución a 425 nm.
3.4.8. Concentración de nitrito
El nitrito (NO2 -) se determina por la formación de un colorante azo púrpura rojizo, producido a pH 2,0 a 2,5 por acoplamiento de sulfanilamida diazotada con diclorhidrato de N- (1-nafftil)-etilendiamida.
Las soluciones necesarias son:
- Reactivo de colorante: se añaden 160 ml de agua a 20ml de ácido fosfórico 85% y 2 g de sulfanilamida. Tras disolver completamente la sulfanilamida, se adicionan 0,2 g de N-(1-naftil)-etilendiamida, se mezcla para favorecer la disolución y se diluye con agua hasta 200 ml. La disolución es estable durante un mes, si se conserva en frigorífico y en un frasco oscuro.
- Solución madre de nitrito (A) de 100 mg l-1 de N(NO2 -). Se pesan 0,49286 g de nitrito sódico cristalizado NO2Na (desecar 1h a 105ºC) y se disuelve en 1000 ml de agua destilada. Se conserva con 1 ml de CHCl3.
- Soluciones intermedia de nitrito (B) de 10 mg l-1. Se toman 100 ml de la solución anterior de 100 mg l-1 (A) y se diluyen hasta 1000 ml con agua destilada.
- Solución patrón (C) de 1 mg l-1 de N(NO2 -). Se toman 100 ml de la solución anterior de 100 mg l-1 (B) y se diluyen hasta 1000 ml con agua destilada para conseguir la solución patrón.
La recta patrón se construye a partir de patrones que van desde 0,05 hasta 1 mg l-1 de N(NO2 -). Se toman 10 ml de cada patrón y del blanco y se agregan 0,4 ml del reactivocolorante. Después de un tiempo de reacción de 10 minutos, se mide la absorbancia de la solución a 543 nm (que presenta un color estable durante 2 horas).
Las muestras se filtran a través de un filtro de 0,22 μm, se preparan las diluciones necesarias hasta un volumen final de 10 ml y se tratan igual que los patrones.
3.4.9. Análisis de amoniaco
Para el análisis del amoniaco en fase gas se empleó un sensor específico de la marca Crowcon (Model GASFLAG, TXGARD-IS). Este sensor da una relación lineal entre la concentración de H2S y el voltaje de respuesta del sensor (medido con voltímetro). En ausencia de NH3, el voltaje es de 40 mV y para concentraciones mayores se puede regular el sensor fijando para cualquier concentración un valor máximo de 430 mV. El coeficiente de regresión lineal (r2) para todas las calibraciones fue mayor de 0,99. Para la calibración más sensible la variación de ±1mV equivale a ±0,26 ppmv de NH3.
3.4.10. Pérdida de carga
Para la medida de la pérdida de carga se utilizaron dos tubos de vidrio conectado cada uno de ellos al biofiltro. Un extremo se conectó a la entrada de gas y el otro a la salida, obteniéndose en una escala graduada la pérdida de carga en milímetros de agua.
3.4.11. Preparación de muestras para microscopía electrónica de barrido
Para obtener imágenes de microscopía electrónica de barrido de los microorganismos, se utilizó un microscopio electrónico FIE Quanta 200 (Philips).
La preparación de la muestra difiere según se encuentren las bacterias en suspensión o adheridas a un soporte. Así para las bacterias en suspensión, se centrifuga la muestra a 7500g durante 10 min a 4ºC, se elimina el sobrenadante y se deposita el pellet sobre un cubreobjetos de 20 mm tratado con polilisina durante 10 min. La polilisina se deposita previamente y se retira el exceso antes de poner la muestra con papel adsorbente. Los cubreobjetos se depositan en un recipiente verticalmente para la fijación con glutaraldehído.
En el caso de las bacterias adheridas en espuma de poliuretano, se toma una pequeña porción de soporte y se elimina la biomasa ocluida con papel adsorbente, a continuación se procede a la fijación con glutaraldehído.
El proceso de fijación con glutaraldehído es idéntico para ambos casos, se sumerge la muestra con una disolución de glutaraldehído al 2,5% en tampón cacodilato de sodio (0,1M, pH 7,2) y se deja actuar durante 1 hora. En la bibliografía hay técnicas similares que en lugar de una 1h de fijación utilizan 12 horas a 4ºC (Varesche et al., 1997); se utilizaron ambas condiciones de fijación y el resultado fue el mismo, por lo que se decidió adoptar el de menor tiempo.
Al cabo de una hora se elimina el glutaraldehído y se añade tampón cacodilato de sodio (0,1M, pH 7,2) realizando 2 lavados de 10 minutos cada uno. La muestra se puede conservar en tampón cacodilato de sodio y a 4ºC hasta su posterior secado hasta punto crítico.
Para realizar el secado hasta punto crítico, se procede a deshidratar la muestra mediante la inmersión en soluciones de concentración creciente de acetona (50%, 70%, 90% y 100%), la duración de cada lavado es de 30 minutos. A continuación, se realiza el secado hasta punto crítico con CO2 para desplazar la acetona y, por último, el metalizado con oro a vacío (condiciones: 15 mA, 120 segundos, distancia 35 mm).
Las muestras una vez tratadas se conservan en campana de secado hasta su posterior observación al microscopio.